

Prion-Like Mechanism in Alzheimer's Disease: Protein misfolding, Seeded Propagation, and Neurodegenerative Overlap (PART 1)
Neurodegenerative diseases (mainly Alzheimer's disease) are molecularly, pathogenically, and neuropathologically considered as protein misfolded disorders characterized by aggregation, oligomer formation and synaptic failure. Classical prion disease involves the conformational conversion of normal prion protein 𝑃𝑟𝑃𝐶 into misfolded forms 𝑃𝑟𝑃𝑆𝑐 ; however, increasing evidence reveals that AD shares a similar prion-like mechanism. Hallmark AD pathologies include amyloid-B Plaques and neuro fibrillary tau tangles exhibit seeded aggregation, oligomer toxicity, and β sheet rich structures similar to prion amyloids. This blog aims to let the viewers examine and decide whether AD and Prion Disease are mechanistically interconnected and to evaluate the role that seed propagation and oligomer formation plays in disease progression.
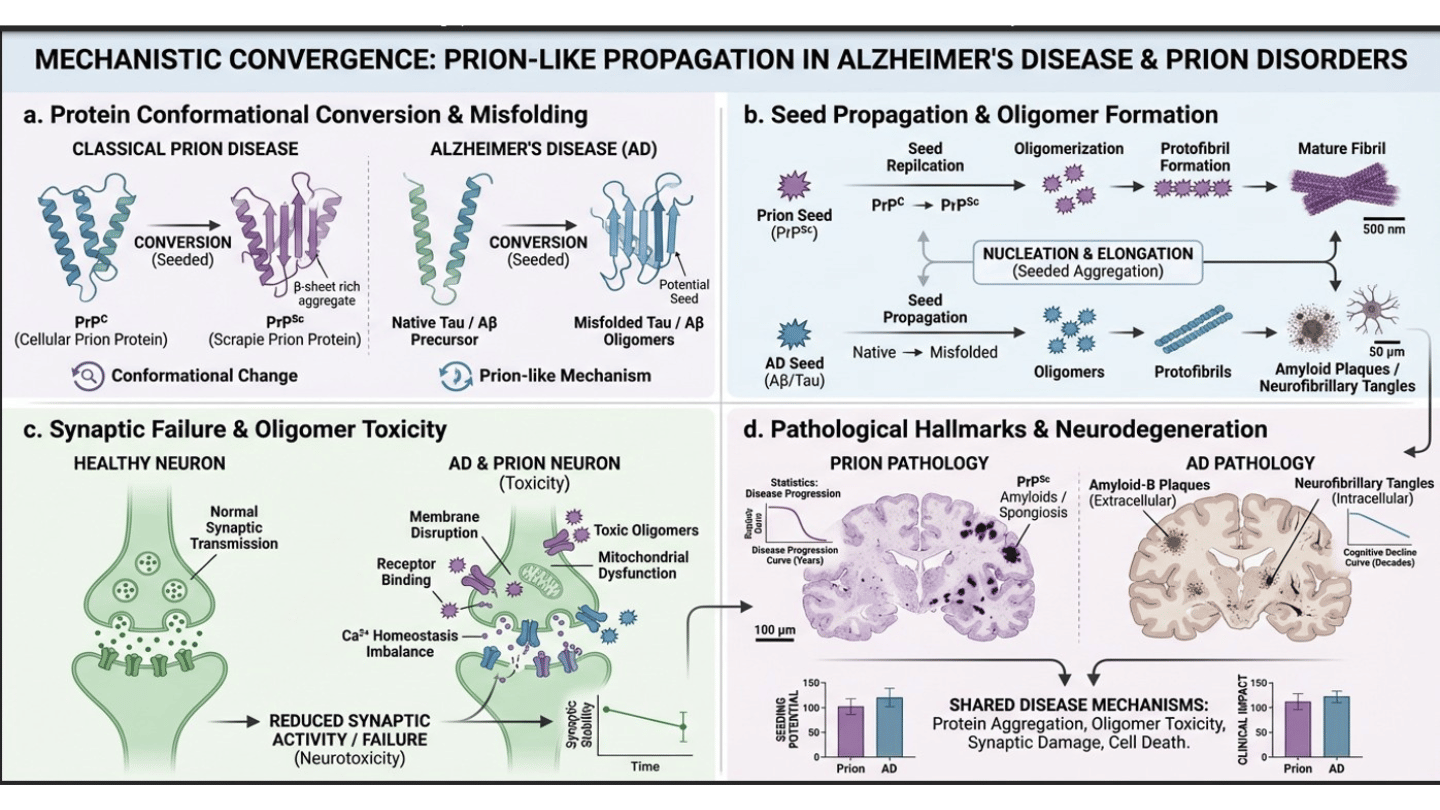
Emerging studies demonstrate that α-syn nuclein, amyloid β, tau act as nucleating particles and exhibit properties similar to that of misfolded forms of prion protein. Soluble Oligomers having seeding like properties act as key pathogenic agents, interacting with PrP Sc leading to synaptic failure and neuronal dysfunction. Prion oligomers constitute structurally diverse intermediates that vary in size, morphology, oligomeric stoichiometry, and conformational organization. Their assembly and propagation follows a nucleation dependent aggregation model where misfolded conformers act as templates in the conversion of native monomers into prion-like assemblies.
Furthermore, it has been revealed that a novel missense mutation (T1071) of PRNP involving the substitution of threonine with isoleucine at codon 107 exhibits diverse clinical phenotypes capable of mimicking Alzheimer's Disease, but biomarker analysis is needed to analyze this further.
Tau and Aβ, as stated exhibit prion-like molecular behavior but AD is not considered infectious between individuals and spread occurs within the brain and not between organisms. Tau is a microtubule related protein, which stabilizes the cytoplasm of neurons. Tau gets hyperphosphorylated and forms neurofibrillary tangles in Alzheimer disease, and may also propagate in a prion-like fashion between cells. Whereas, Ab (amyloid-beta) is a polypeptide that is formed out of amyloid precursor protein (APP). Aβ misfolds and aggregates into oligomers and plaques in Alzheimer disease, which also contributes to neurotoxicity and may also exhibit prion seeding.
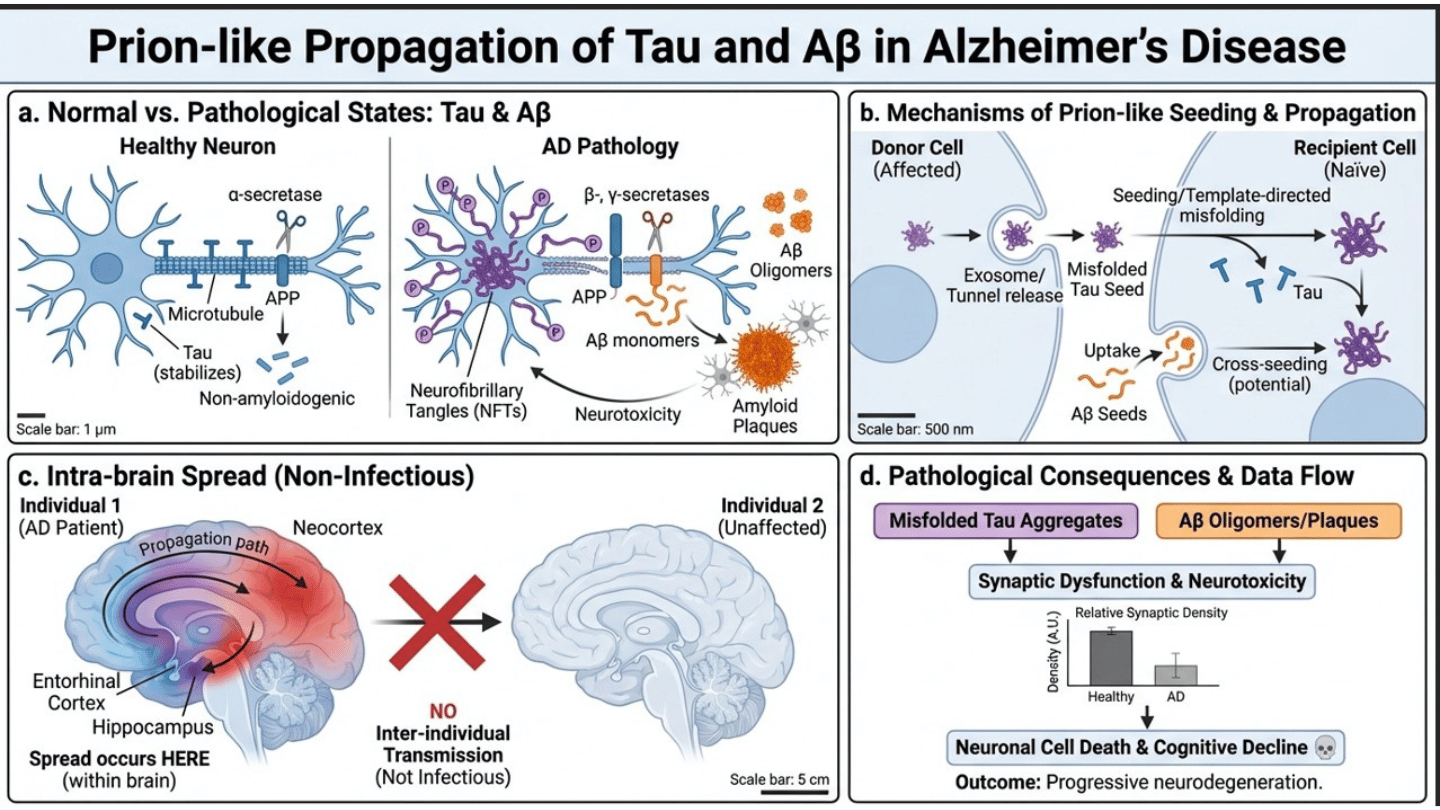
Although Aβ Seed propagation pathology in the brain is unknown, structural and experimental analysis provides mechanistic insight into the protein aggregation pathway and the concept of Prion-like proteins in AD. Understanding the molecular mechanisms may open a new pathway for therapeutic interventions. Targeting oligomer formation, blocking protein production, preventing aggregation and disrupting Aβ-Tau interactions are considered promising strategies in preventing both prion-like diseases and AD.
Looking at the information stated above, are prion diseases and Alzheimer diseases essentially the same disease or are they different conditions associated only by pathogenic processes that are prion-like in nature?
{kind=link}