

Molecular Docking:
A computational technique called molecular docking is used to predict the interactions between two structures, like a protein and ligand, protein and nucleotide, and even two proteins. The process aims to determine the best possible match between two molecules and establish the most stable orientation when two molecules combine to form a complex with the lowest overall energy. Various interaction types are considered in this procedure, such as hydrogen bonds, coulombic interactions, van der Waals forces, and electrostatic forces.
Types of Molecular Docking:
Targeted or site-specific docking: Targeted docking is a molecular docking approach focused on a known specific region of the protein, typically the active site or another site of known biological significance.
Blind docking: Blind docking is a type of molecular docking approach where the entire surface of the protein is considered for potential binding sites. This method is used when the exact binding site is unknown or when multiple potential binding sites need to be explored.
How can we perform molecular docking?
Well, there are two ways of performing molecular docking:
1. Standalone molecular docking
2. By using webservers
Standalone molecular Docking:
1.Program installation: First and foremost, it's crucial to download essential tools to your computer. In my experience, Discovery Studio, MGL Tools, and AutoDock Vina are the top choices. These programs offer easy docking processes and are incredibly user-friendly.
a. Autodock Vina: After downloading Autodock Vena, you will have access to two application tools and a license file. Vina application is the main executable of AutoDock Vina and performs molecular docking. You run this application to carry out docking calculations, obtaining binding modes and affinity estimates. Additionally, Vina Split takes the combined output file from a Vina docking run, which contains multiple poses, and splits it into individual files, each representing a single binding pose
b. Open Babbel: I strongly recommend downloading Open Babel to your computer. This program is essential for changing file formats when using certain docking programs. Open Babel is incredibly user-friendly and will be very helpful for your needs.
c. Folder Preparation: For those who may not be very familiar with computer systems, I suggest creating a dedicated folder for all docking-related files, structures, and Autodock Vina applications and the license when performing molecular docking.
2. Protein Preparation:
a. Download Protein Structures: Make sure to download the most recent protein structure from the RCSB PDB database. Select the structure that has been determined through X-ray crystallography with a wavelength of less than 4 Angstroms, indicating a resolution of less than 4 Angstroms. It is essential to locate the protein chain, for this check the macromolecule section of the RCSB PDB database. The structure should be downloaded in PDB format.
b. Structure Prediction: If the structure you need is not available in the RCSB PDB database, you can predict the structure by utilizing tools such as AlphaFold2, I-Tasser, and Phyre 2.
c. Protein Cleaning and Chain Selection: Employ Discovery Studio to select and prepare the necessary protein chain by eliminating water molecules, ligands, and surplus chains. Following this, save the file in PDB format.
d.Add charge and Polar Hydrogens: Open the recently saved PDB file in AutoDock Tools. Add polar hydrogens and Kowlman charge.

3. Active site prediction: In targeted docking, the active site is predetermined before docking. The most effective approach is to thoroughly review the literature for the selection of active site amino acid residues or utilize online tools such as CASTp or DoGSite Scorer. It is advisable to employ three to four tools for active site prediction, and then proceed with the common amino acid residue results. select the active site amino acid residues in the Autodock Tools program. Choose, select, and save the Protein structure with the active site in the designated docking folder in PDBQT format.

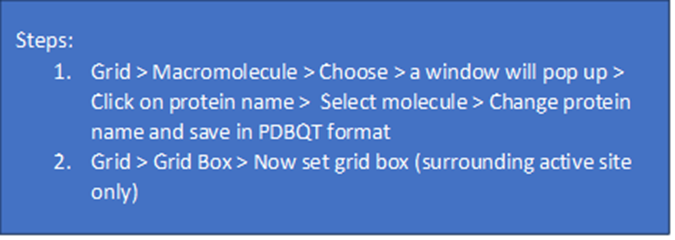
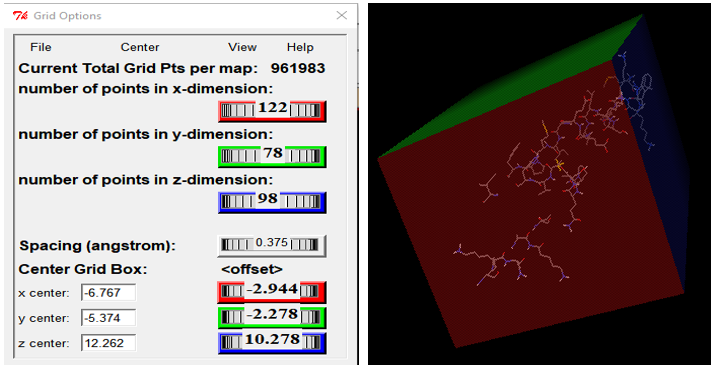
4. Grid Box Formation: The grid box establishes the boundary for the docking process. When conducting targeted docking, the grid box will exclusively surround the active site, in contrast to blind docking, where the entire macromolecule is encompassed within the grid box. In the MGL tool program, set the size and dimensions of the grid box. Remember to capture a screenshot of the grid box dimensions, as these dimensions are essential for the subsequent stages of the process.
5. Ligand Preparation: The selection of a ligand for use as a drug should be based on the ADMET criteria. Download Ligand structure in SDF or PDB format from Chambel, Pubchem, or Zinc databases. Open ligand structure in the Autodock tools program and add polar hydrogens and charges. Save Ligand Structure in PDBQT format.
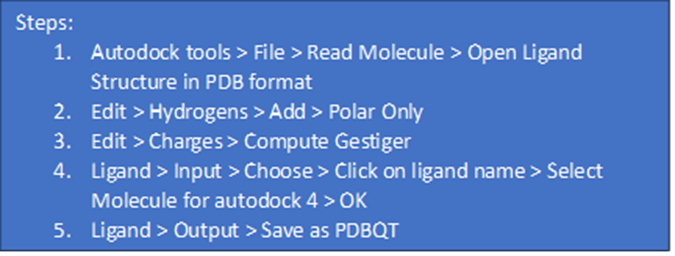
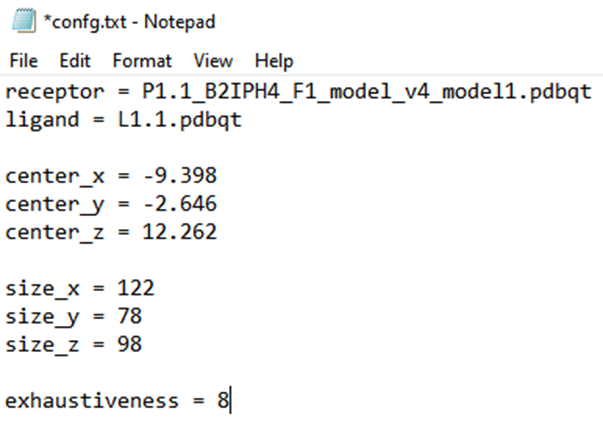
6. Configuration file format and preparation: To prepare the configuration file, open Notepad and create a .txt file. Insert the receptor and ligand names in PDBQT format, along with the grid box size and dimension information. Save this file in the designated docking folder.
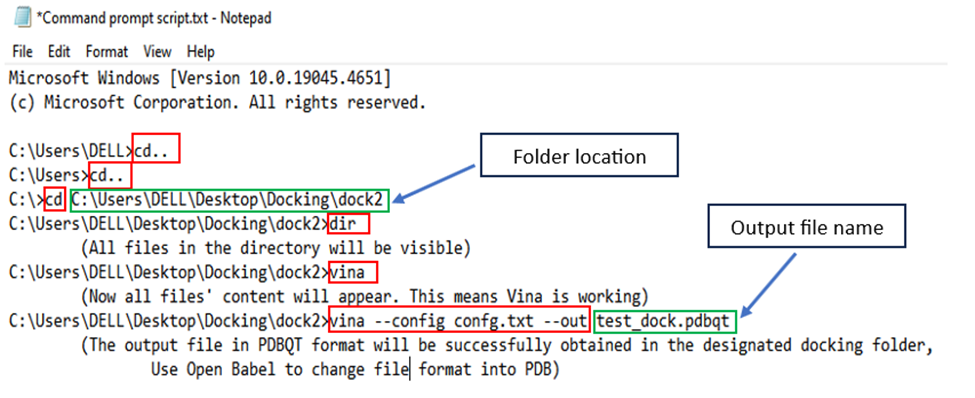
7. Command Prompt System: Open the command prompt system and add the following commands.
8. Results Analysis: You will obtain a total of 9 results and select only those with the lowest binding energy. A more negative score indicates a stable complex. Prefer results with an RMSD value of less than 2, as a lower RMSD indicates a better model.

9. Docking Analysis: Open the protein file in PDB format in Discovery Studio and also open the output file in PDB format. After that, select the first ligand and copy its structure using the Ctrl + C function. Next, paste this ligand structure using Ctrl + V in the protein structure window of Discovery Studio. Finally, click on the 2D structure in the sidebar and observe the interactions and bonds between ligands and residues.
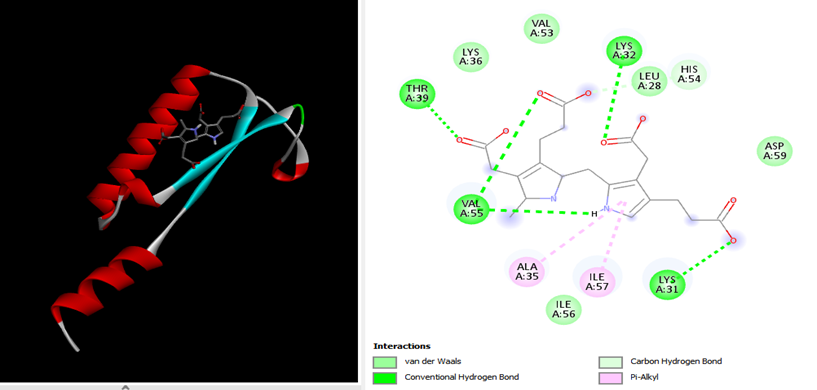
Two essential stages in standalone and web server docking include:
Initially, the program looks into the conformational space and displays potential locations for the molecule to be bound to the target. Then, the program calculates the energy values for each potential linkage.
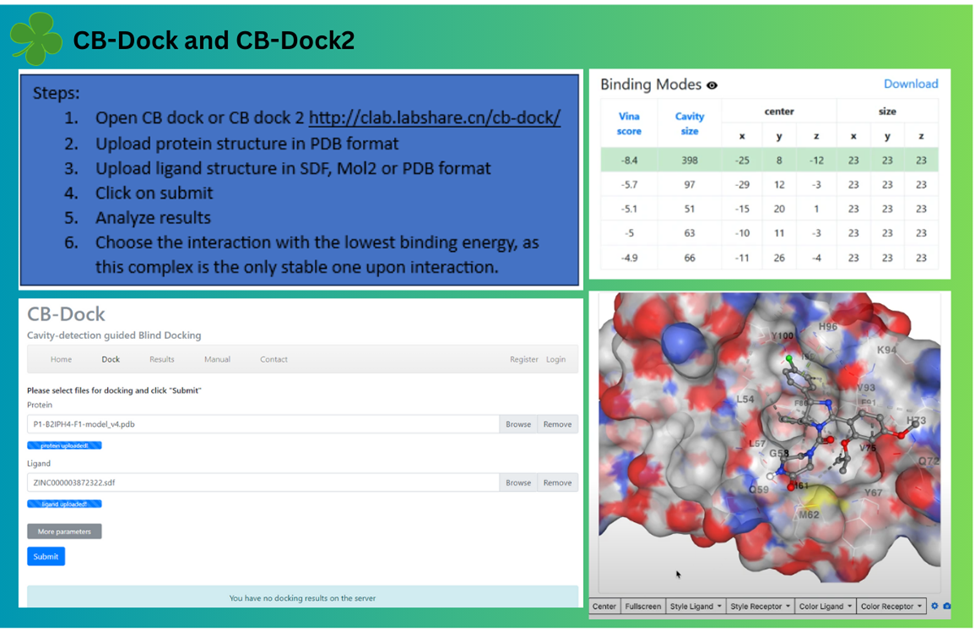
Docking via webservers: For docking via webservers one can use CB-dock and CB-dock2. CB-Dock is a user-friendly web server developed by Yang Cao lab, and it, along with its successor CB-Dock2, is considered one of the best online tools for performing protein-ligand blind molecular docking. CB-Dock utilizes AutoDock Vina for docking
Applications of Molecular Docking:
Molecular docking simulations streamline the drug development process, significantly reducing time and costs while delivering precise results. By molecular docking, evaluating the interactions between a drug and its receptor becomes very easy. These simulations identify multiple binding possibilities within the target molecule's groove and determine the optimal orientation for a ligand. By employing docking with a scoring function, large drug databases can be swiftly screened in silico to identify compounds with a high probability of binding to specific protein targets.
Conclusion:
Molecular docking stands as a vital computational approach for drug development, accurately predicting the interaction between ligands and proteins. Despite its complexity and scoring intricacies, molecular docking remains invaluable in the early stages of drug development due to its effectiveness, efficiency, and reduced time and labor requirements.

{kind=link}