

A Comprehensive Guide to Multiple Sequence Alignment Using ClustalW: From Sequence Extraction to Phylogenetic Analysis
Multiple Sequence Alignment can be seen as generalization of Pairwise Sequence Alignment. Instead of aligning two sequences, number of sequences can be sequenced simultaneously where n>2.
MSA is obtained by inserting gaps (“-”) into sequences such that the resulting sequences have all length L and can be arranged in a matrix of rows and L columns, where each column represents a homologous position.
Uses:
- MSA can help to develop a sequence “finger print” which allows the identification of members of distantly related protein family (motif).
- MSA can help us to reveal biological facts about proteins (e.g. how protein function has changed or evolutionary pressure acting on a gene).
- To establish homology for phylogenetic analysis.
- To identify primers and probes to search for homologous sequences in other organisms.
Crucial points for Genome Sequencing:
- Random fragments of a large molecule are sequenced and those that overlap are found by multiple sequence alignment program.
- Sequence may be from one strand of the DNA or other, so complements of each sequence must also be compared.
- All the overlapping pairs of sequence fragments must be assembled into large composite genome sequence.
Multiple Sequence Alignment using ClustalW:
Open UniProt and enter query (e.g. IL6) in the search bar then press search.
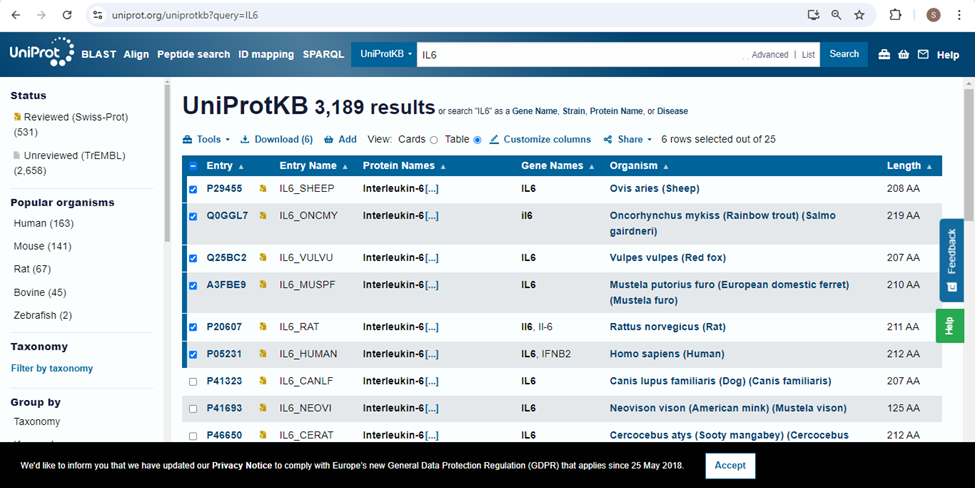
Select the 6 sequences and download the sequences in FASTA (complete sequence) format.
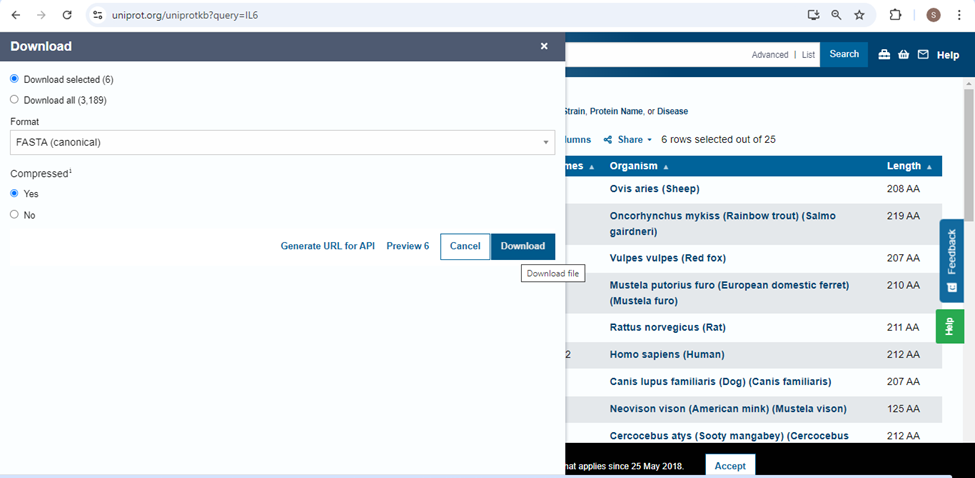
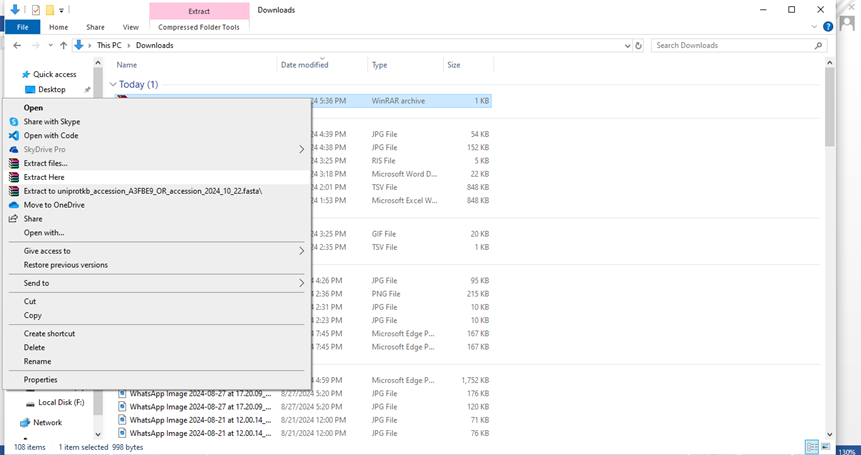
- The file will be downloaded as ZIP file. Go to the folder where file is downloaded, right click on it and the click on “Extract here”.
Open the ClutalW tool and upload the downloaded sequence file and then click on “execute multiple sequence alignment”. the results will show the multiple sequence alignment of the selected sequences.
- To construct a phylogram of the result, scroll to the bottom of the results page and in the “select tree menu”, click on “PhyML” and then click “Exec” to execute this process.
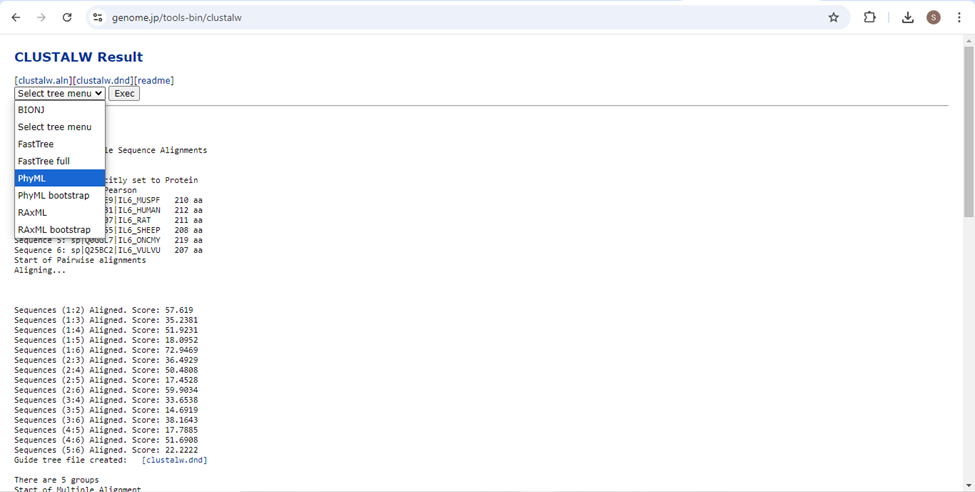
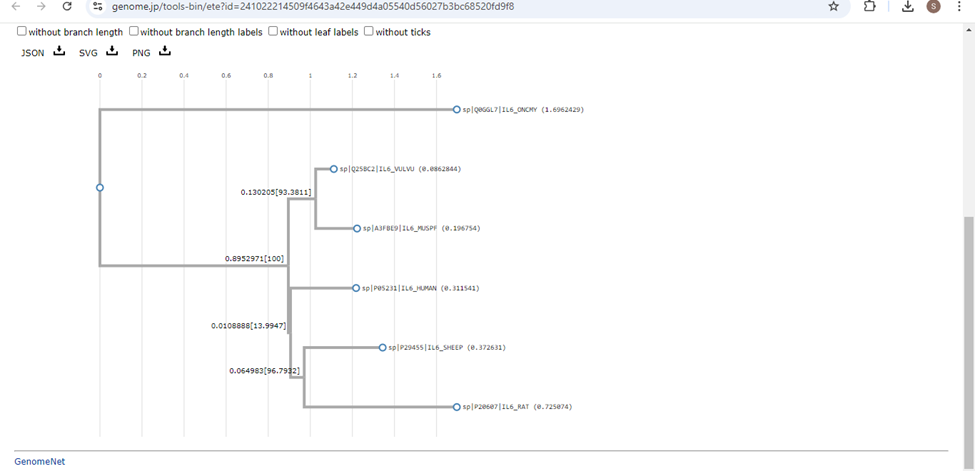
- The Phylogram will be shown as a result. You can save the phylogram in PNG, SVG or JSON.
Interpretation:
- In the MSAscoringscheme, a penalty is subtracted for each gap introduced into an alignment because the gap increases uncertainty into an alignment.
- The gap penalty is used to help decide whether or not to accept a gap or insertion in an alignment.
- Biologically, it should in general be easier for a sequence to accept a different residue in a position, rather than having parts of the sequence chopped away or inserted. Gaps/insertions should therefore be morerarethan point mutations (substitutions).
In general, the lower the gapping penalties, the more gaps and more identities are detected but this should be considered in relation to biological significance.
References:1. GenomeNet ClustalW Information. "CLUSTALW - Multiple Sequence Alignment Program." GenomeNet. Accessed on October 27, 2024. https://www.genome.jp/tools-bin/clustalw
2. Münster University Presentation on ClustalW. "PowerPoint Presentation on Multiple Sequence Alignment and Phylogeny." University of Münster. Accessed on October 27, 2024. https://www.fh-muenster.de

{kind=link}